Jul 6 2016
With the summer fun and flip flop season in full swing, those suffering from discolored toenails start to worry about what others think of their feet. Toenail fungus sufferers can now enjoy clear nail beds with the FDA 510(k) approval of the Lunula Laser, an Erchonia Corporation low-level laser.
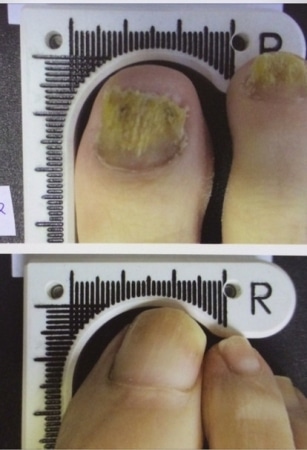 Toenails before and after receiving Lunula treatment. (PRNewsFoto/Erchonia Corporation)
Toenails before and after receiving Lunula treatment. (PRNewsFoto/Erchonia Corporation)
The Lunula device increases the amount of clear nail in patients infected with onychomycosis, or nail fungus. In the clinical trial, 67 percent of patients met the success criteria of three millimeters of clear nail growth. By six months after the initial treatment, these patients average more than five millimeters of new growth.
This is the first and only low-level laser to receive FDA 510(k) marketing clearance for onychomycosis.
"Lunula is the future of treating onychomycosis," said Dr. Kerry Zang, founder of the Arizona Institute of Footcare. "The results seen in the clinical trial are spectacular and we are eager to provide Americans with the opportunity to treat this nail fungus with an effective product."
The portable Lunula device applies a laser to the area infected by onychomycosis. During the study, patients between 18 and 70 years old received treatments once a week for four weeks.
Previously, treatment for nail fungus included prescription oral antifungal medication, which increased potential for liver toxicity issues, or else called for ineffective, over-the-counter topical creams.
"Nail fungus is a big problem and the toxicity of the available drugs is almost as bad," said Dr. Robert Sullivan. "Lunula gives doctors a viable treatment option: no blood tests, no pain and no mess."
Zang and Sullivan lead the research behind Lunula. Prior to FDA 510(k) approval, the product went through four clinical trials which recorded no known side effects.
Preorders begin June 30, with delivery scheduled for mid-July.
The 510(k) is a premarketing submission to the FDA that demonstrates that the device marketed is safe and effective. The premarket approval for the 510(k) is the most rigorous type of device marketing application accepted by the FDA.